Dataset revolutionizes understanding of beta barrels, promising targets for vaccine development
LAWRENCE — Examples of infections tied to gram-negative bacteria include pneumonia, bloodstream infections, wound-site infections and meningitis, according to the Centers for Disease Control and Prevention.
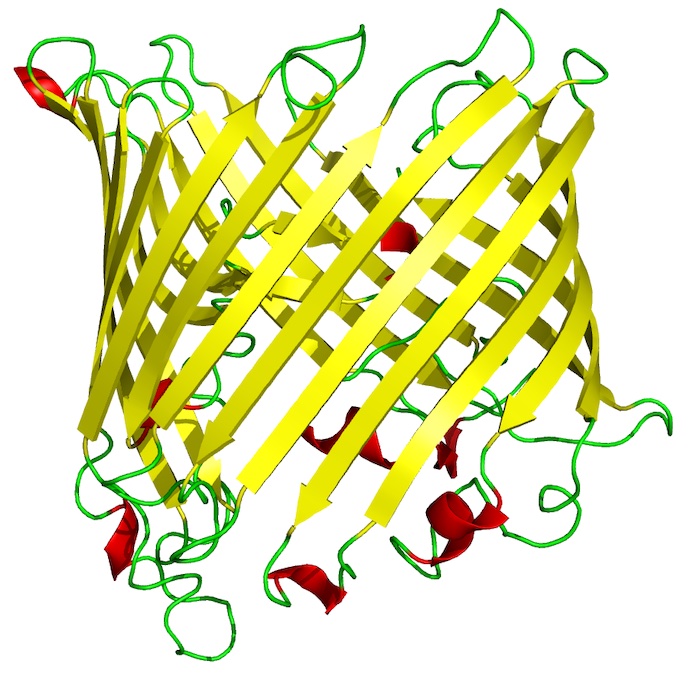
Scientists have focused on tubelike “outer-membrane proteins” found in certain types of gram-negative bacteria that could be crucial to developing vaccines against a range of such infections, some of which show increasing drug resistance. These outer-membrane proteins often assume a tubelike shape, known as beta barrels, that have great potential as vaccine targets.
Now, researchers from the University of Kansas have created a new and powerful dataset shedding light on different types of beta barrels as well as their evolutionary relationships in order to facilitate drug development. Their findings recently were published in the peer-reviewed journal Proceedings of the National Academy of Sciences.
According to co-author Joanna Slusky, associate professor of molecular biosciences and computational biology at KU who oversaw the research in her lab, the new understanding of beta barrels enables fresh lines of scientific inquiry as well as drug development.

“Outer membrane proteins reside in the outer membrane of gram-negative bacteria, and all of these membrane proteins take on a beta-barrel structure, sharing a common basic topology.” Slusky said. “The outer membrane plays essential roles, including nutrient import, toxin export, adhesion, enzymatic activity and environmental adaptation, all governed by these outer membrane proteins.”
The Slusky team’s approach to producing the new dataset diverged from past efforts, which only sought to catalog relatives of known outer-membrane proteins. The new method, dubbed “IsItABarrel,” has revealed more than 270,000 previously unidentified outer-membrane proteins that could interest to vaccine researchers. Slusky’s group has posted the database online to enable such work.
“Recognizing their shared beta barrel characteristic as a distinctive shape, we developed an algorithm that yielded approximately 1.9 million instances of these proteins,” Slusky said. “From there, we cluster them into distinct groups. The predominant group accounts for around 1.4 million instances. Then, a substantial portion, approximately 500,000 instances, falls into various other groups. This suggests that nature independently developed this fold multiple times."
Whereas the Slusky lab and others had previously detected two or three instances where protein evolution had converged on the beta barrel shape, now the KU team has identified 11 independent instances of this occurrence in various bacterial types.
“Our initial exploration revealed more bacteria had this type of protein than anticipated, including those previously underrepresented,” Slusky said. “Furthermore, we observed an enhanced presence of transmembrane beta barrels in bacteria we already knew had these proteins. Our confidence metrics demonstrate minimal false positives, as well as comparable false negatives to other methods. This affirms the reliability of our algorithm.”
The Slusky Lab’s investigation also revealed that while many proteins show the characteristic “barrel signal,” or sequences of amino acids long known to fold into the barrel shape, other types of proteins feature barrel signs that were unknown until the team’s analysis with “IsItABarrel.”
“This sequence motif plays a pivotal role in facilitating both the insertion and folding of proteins within the membrane,” said the KU researcher. “The commonly recognized motif is exclusively present in the prototypical beta barrels. Conversely, alternative proteins exhibit entirely different motifs that lead to the formation of distinct barrel groups within the membrane. This discovery provides additional substantiation for the concept of independent evolution. However, it also underscores the limitations in our comprehension of the folding process for these other proteins.”
Slusky said the discovery that bacteria can make beta barrels using new sequence motifs of amino acids should provoke many new lines of research.
“Do they use the same folding mechanism as the aforementioned proteins, or do they employ an entirely different approach?” she said. “It remains uncertain whether these proteins use a similar mechanism for insertion, potentially with the assistance of a distinct protein. Alternatively, their insertion process may be fundamentally distinct. Definitive answers to these inquiries are still pending."
Slusky’s collaborators on the work were lead author Daniel Montezano, a KU postdoctoral researcher, along with co-authors Rebecca Bernstein, a high school researcher, and Matthew Copeland, research engineer with KU’s Computational Biology Program.
Photo: Image of an 18-strand beta barrel from S. typhimurium that allows sucrose to diffuse through a membrane. Credit: Wikimedia